
La recherche académique
Lorsque dans une famille, est identifiée une transmission autosomique dominante de la maladie d'Alzheimer avec un âge de début avant 65 ans, la probabilité d'identifier une cause connue et validée est entre 83 et 86 %. Donc 17% des formes autosomiques dominantes restent inexpliquées sur le plan génétique. Le CNRMAJ a mis en place différentes stratégies de recherche destinées à identifier de nouvelles causes génétiques dans ces familles restées "négatives".
Les investigations menées recherchent 2 types d'anomalies : (i) les variations ponctuelles ou mutations (ii) les variations du nombre de copie de gène (à l'instar des duplications d'APP). Pour parvenir à relever ce défi, il est capital de suivre une stratégie que l'on qualifie "sans a priori". Cela veut dire que les investigations de recherche étudient l'ensemble du génome. Les techniques que nous employons sont d'une part le séquençage à haut-débit de l'exome (c.a.d la partie de notre génome codant pour des protéines) et d'autre part la CGH Array (Comparative Genomic Hybridization Array).
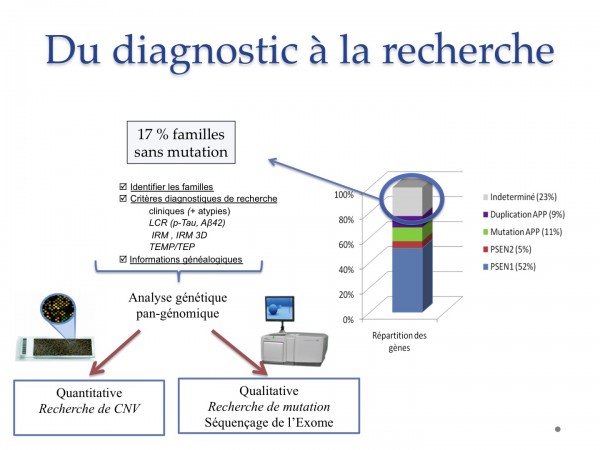
Stratégies de recherches pour les familles sans anomalies génétiques identifiées
Stratégie N°1 : recherche d'anomalie quantitative par CGH Array
Dans cette première stratégie, l'idée est de rechercher des variations du nombre de copie de gène, là encore sans a priori. Les duplications d'APP sont un exemple de ces variations (en l'occurrence une augmentation) du nombre de copies du gène APP dans le génome. Nous avons naturellement 2 copies de chaque gène situé sur un chromosome issu du père et de la mère. Cependant dans certaines conditions, il y a copie supplémentaire que l'on appelle duplication ou à l'inverse perte d'une copie, alors appelée délétion. Ces variations du nombre de copie (CNV pour copy number variant) peuvent aller d'un nucléotide jusqu'à plusieurs gènes.
L'identification de ces CNV est rendue possible par la CGH Array. Ainsi en colorant par fluorochrome vert l'ADN d'un contrôle sain et par fluorochrome rouge l'ADN du patient, puis après mélange des échantillons, coupures des ADN en petits morceaux et fixation (appelé hybridation) de ces morceaux sur des sondes collées à une plaque de verre, on révèle la couleur de la sonde.
Si les quantités d'ADN de patient et de contrôle sont les mêmes, la sonde brillera en vert+rouge => jaune. C'est la situation normale. Si l'ADN de patient a subi une délétion, il y en aura moins que le contrôle, la sonde apparaitra en vert. A l'inverse, si il y a eu duplication, l'excès d'ADN de patient rendra la sonde fluorescente en rouge. Il ne reste plus qu'à visualiser toutes les sondes posées sur la plaque de verre avec un scanner ultra-sensible en connaissant exactement à quels portions du génome correspond chaque sonde. On est ainsi capable de dire précisément quelle partie du génome du patient a subit une délétion ou duplication.
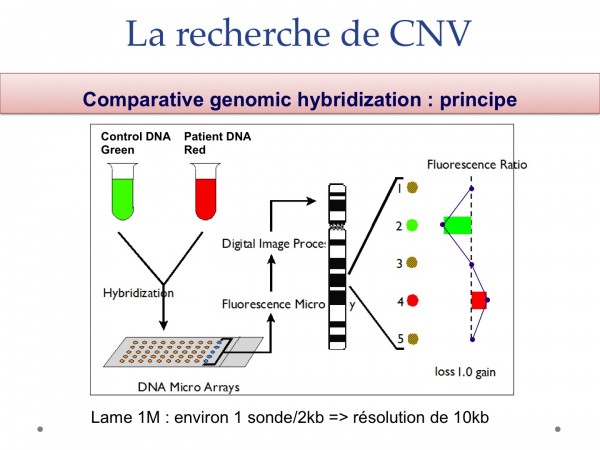
Recherche de variation du nombre de copie de gène(s) par CGH
Cette seconde stratégie est également efficace puisqu'elle a permis au CNRMAJ d'identifier des CNV rares mais intéressant des gènes importants vis à vis de la maladie d'Alzheimer. La rareté de ces variations impose de rester prudent sur la pathogénicité mais nous pouvons envisager que d'autres équipes objectiveront certains de ces gènes dans leurs familles renforçant la certitude concernant ces découvertes.

L'idée, dans ce cas, est d'identifier N familles (5 dans l'exemple ci-dessous) toutes négatives pour les gènes connus responsables de formes autosomiques dominantes. Plus le nombre sera grand et plus la stratégie aura une chance d'aboutir.
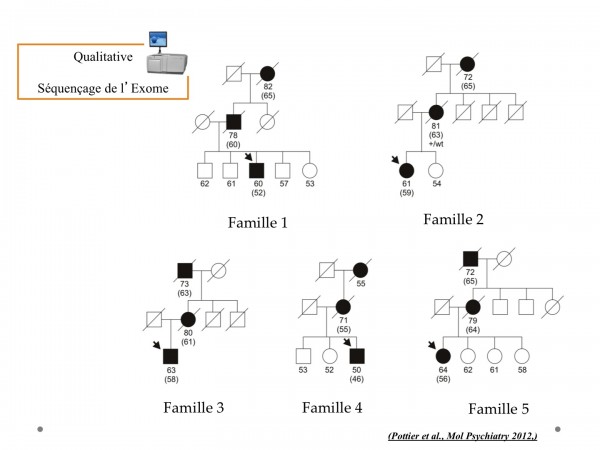
Une fois les séquençages de l'exome réalisés chez tous les cas index (marqué d'une flèche), les mutations identifiées pour chacun des individus seront comparés dans les N familles afin de retrouver au moins un gène en commun. Dans l'exemple ci-dessous, on pouvait espérer retrouver une gène en commun pour 3, 4 voire les 5 familles. Cela présuppose que le phénotype de chaque patient et dans chaque famille ait été particulièrement étudié afin de garantir que le groupe est bien homogène avant d'envisager une cause génétique commune.
Cette stratégie est tout à fait efficace et a permis au CNRMAJ d'identifier un premier gène potentiellement causal : SORL1 dont la validation est en cours actuellement.
